This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison.

Crouzon Syndrome is a genetic disorder characterized by craniosynostosis, the premature fusion of skull joints in infants (1). It affects about 16.5 people in every million, and it is responsible for almost 5% of all cases of craniosynostosis (2). Genetics Home Reference states Crouzon Syndrome is an autosomal dominant disease, and, therefore, inherited by a single parent with the condition or acquired due to a new mutation in one allele. Symptoms of craniosynostosis include abnormal shape of the head and face (1). This can lead to intercranial pressure which often results in poor vision and headaches. Poor head development also leads to deafness and inner ear problems in some patients. Crouzon patients also often experience abnormal development of the trachea and upper airways causing airway obstruction and nasopharyngeal narrowing. In a small number of cases Crouzon patients also experience decreased mental abilities. Due to the wide range and varied nature of abnormal development problems, each patient requires a personalized medical care plan. Treatments are often required to aid in respiration, prevent visual impairment, reduce hearing and/or monitor speech. Surgical procedures are common treatments among these patients and include shunting to relieve water on the brain and tracheostomies to reduce airway complications (3). According to Crouzonsyndrome.net, surgery may also be used to reshape the skull, eye sockets, and/or jaw for a more normal appearance.
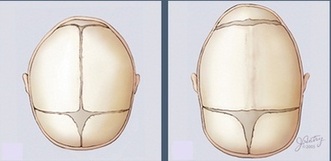
Left - normal skull plate fusion, Right - craniosynostosis
|
The cause of Crouzon syndrome is a mutation in the fgfr2 gene which codes for fibroblast growth factor receptor 2 and lies on human chromosome 10 (4). Mutations in this gene have been linked to several disorders involving craniosynostosis including Crouzon Syndrome (5). Amongst craniosynostosis syndromes fgfr2 mutations are extremely common, but there are few known genotype-phenotype correlations. In some cases, different individual mutations have been shown to cause the same disorder, while in others, identical mutations have resulted in different conditions(6). If fact, Genetics Home Reference, states that more than 35 different mutations in this gene have been shown to result in Crouzon Syndrome. According to that same source, most of the mutations are single-nucleotide substitutions, but some are insertions or deletions. These mutations are gain-of-function, and act by overstimulating the FGFR2 protein which causes the premature skull plate fusions discussed above.
|
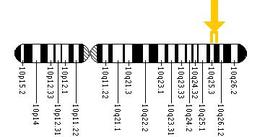
Figure 2. Fgfr2 on chromosome 10
|
|
[1] Seattle Children's Hospital. Bone, Joint, and Muscle Conditions: Crouzon Syndrome. Retrieved 2/2/2012. http://craniofacial.seattlechildrens.org/medical-conditions/bone-joint-muscle-conditions/crouzon
[2] Cohen Jr MM, Kreiborg S. Birth prevalence studies of the Crouzon syndrome: comparison of direct and indirect methods (1992) Clinical Genetics, 41: 12–15. DOI: 10.1111/j.1399-0004.1992.tb03620.x [3] Chen, H., MD, MS, FAAP, FACMG. Medscape Reference: Genetics of Crouzon Syndrome Treatment and Management. Retrieved 2/7/2012. http://emedicine.medscape.com/article/942989-treatment#a1128 [4] National Center for Biotechnology Information. FGFR2 fibroblast growth factor receptor 2 [ Homo sapiens ]. Retrieved 2/7/2012. http://www.ncbi.nlm.nih.gov/gene/2263 [5] Robin, N. H., Falk, M. J., Haldeman-Englert, C. R. FGFR-Related Craniosynostosis Syndromes. Last updated: 2011 PMID: 20301628 [6] van Ravenswaaij-Arts CM, van den Ouweland AM, Hoogeboom AJ, Herbergs J, Pals G. (2002) From gene to disease; craniosynostosis syndromes due to FGFR2-mutation Ned Tijdschr Geneeskd, Jan 12;146(2):63-6. PMID: 11820058 |
|
